
ASHG 2023 conference: Exclusive insights from Complete Genomics

Nov 14, 2023
At the American Society for Human Genetics in Washington D.C., geneticists worldwide were briefed on Complete Genomics’ DNBSEQ revolutionizing full genome sequencing, the use of DNBSEQ-T7 in clinical labs for structural variant detection, and a University of Massachusetts researcher’s use of STOmics’s Stereo-Seq and DNBSEQ-T7 data.
DNBSEQ technology and new advances in whole genome sequencing

Dr. Radoje Drmanac, Phd
Chief Scientific Officer, Complete Genomics
Key takeaways
- End-to-End genomic solutions: Complete Genomics provides comprehensive genomic research solutions, from sample processing to bioinformatics.
- DNBSEQ technology: DNBSEQ line, including DNBSEQ-G99 and DNBSEQ-T7, offers high-throughput, accurate genome sequencing.
- Cost-effective genome sequencing: Notable reductions in sequencing costs, exemplified by the $150 genome milestone achieved with DNBSEQ-T7.
Recording
Talk summary
Dr. Drmanac introduced Complete Genomics, founded in 2005 in Silicon Valley and acquired in 2013. The company, with over $1B in investments and technology development, now boasts over 200 employees, nearly 600 patents, around 3,000 global customers, and more than 6,000 research publications.
Regarding these publications, the vast majority are RNA-Seq findings, and a long tail of additional techniques (WGS, targeted sequencing, metagenomics…) typical for next-generation sequencing. Complete Genomics has paid attention to the entire laboratory workflow, offering liquid handling automation from sample disruption and handling, to nucleic acid purification, automated NGS library preparation, sequencing, all the way through to bioinformatics solutions.
The DNBSEQ method (DNA NanoBall sequencing) forms the common technology throughout the sequencer lineup. Their “G-Series” DNBSEQ-G99 produces 8 to 48 GB of sequence per run; and can be set up to finish a run “in a few hours if needed” for time-sensitive applications. The “T-Series” DNBSEQ-T7 produces 1-7 Tb of sequence per run, with a four-flowcell system, which is unique and offers great flexibility in the life sciences industry.
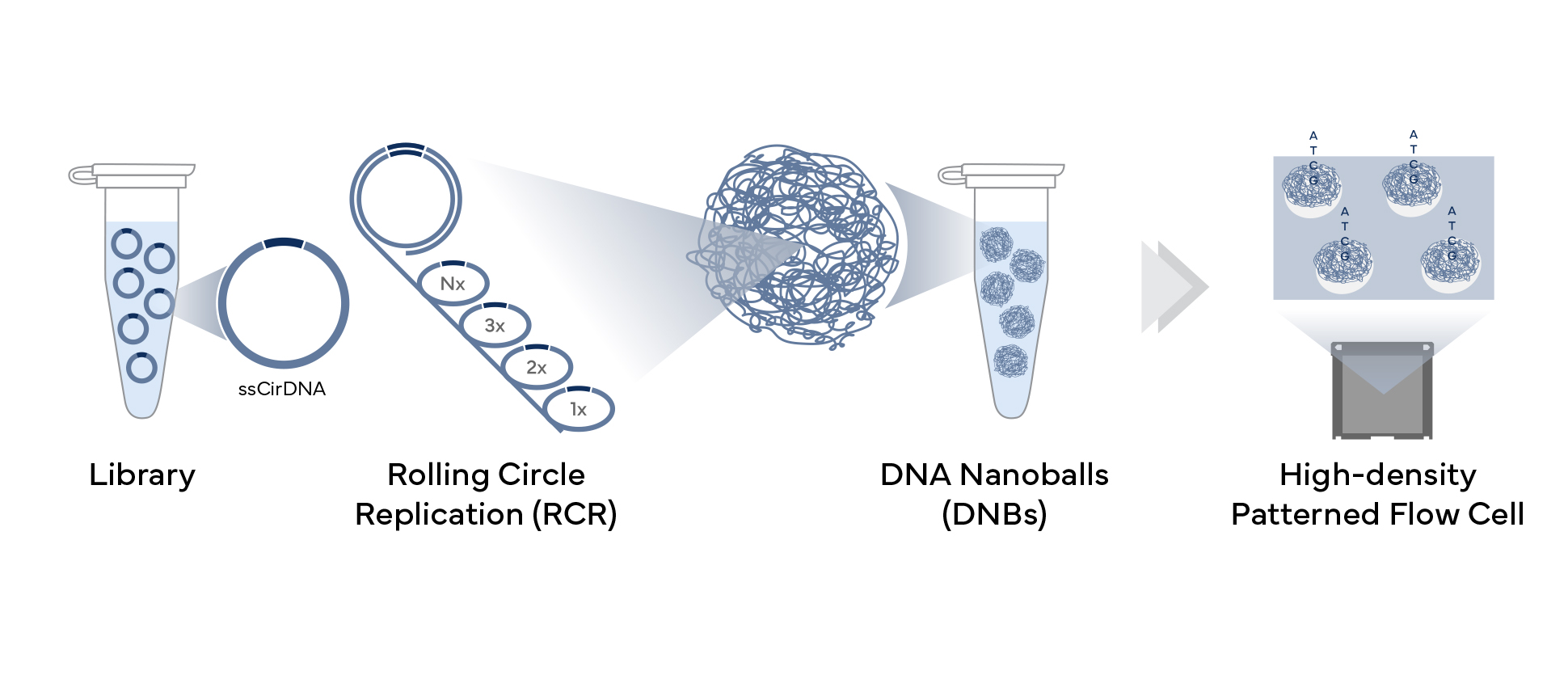
Dr. Drmanac then presented the DNBSEQ technology, illustrated in Figure 1. Rolling circle replication uses the same single template repeatedly, which is an important nuance of the technology: there are no clonal errors that arise, because only the original template is read. About two hundred copies of the template comprise the DNA NanoBalls (DNBs). A high-density, patterned flowcell contains a single DNB at >95% occupancy.
Another important benefit of not having clonal amplification errors is the elimination of index hopping, a known phenomenon with amplification-based template preparation.
He then presented a timeline of whole genome sequencing costs. Starting with a $50,000 genome in 2009 and the $5,000 genome in 2010, the timeline continued with a series of NGS instruments launched including the DNBSEQ-G400 in 2018, and the $150 genome with the DNBSEQ-T7 in 2019.
To illustrate what increases in density (and throughput) are possible, he showed the current T20 flowcell format: DNBs about 200nm in size, and 500nm apart (called “pitch size”). A future Txx model flowcell could have the DNBs in the same size, at only 250nm pitch size (raising the density by 400%). With only 125nm pitch size and DNBs shrunk to only 100nm in size, another 400% (four-fold) increase could be achieved.
Shifting to the applications of whole-genome sequencing, he mentioned the 2014 Nature publication1 of the diagnosis of severe intellectual disability of 50 individuals.
Dr. Drmanac continued with a brief description of indel and SNV accuracy of the whole-genome sequence of NA12878, with PCR-free libraries independently prepared in duplicate and run on the DNBSEQ-G400. Discordant data between replicates were between 2.05% – 2.65%. Of interest was a side-by-side comparison chart of Genome in a Bottle (GIAB) sample HG002 between the DNBSEQ-G400 and the Illumina NovaSeq 6000Ô. For single nucleotide variants (SNVs), the false negative (FN), false positive (FP), recall and precision were all comparable. In contrast, for indels the FN and FP were improved by two-fold, depending on the analysis pipeline used (whether DNAScope or GATK).
In a different analyses with the same sample and datasets (specifically HG002 and 30x coverage, both using PCR-free libraries) the NIST “Challenging Medically Relevant Gene” benchmark curates 273 genes, and the Complete Genomics measures across the board were essentially equivalent. Of the 273 genes, Illumina could detect 127 of these genes perfectly, while Complete Genomics could detect 130.
Dr. Drmanac concluded by presenting single-tube Long Fragment Read technology (stLFR) for haplotype phasing, and a new technology called Spatio-Temporal Enhanced REsolution Omic Sequencing (Stereo-Seq) for spatial transcriptomics. For long fragment reads, a single-tube protocol enables 70Mb contig lengths (N50), with the ability to detect compound heterozygote SNVs, offering higher coverage of difficult regions such as tandem repeats or the aforementioned challenging medically related genes and areas of poor coverage termed “blind zones” in genes such as SMN1, NBPF4 and CHRNA7.
Rare disease diagnostics through WGS, Optical Mapping and Transcriptomics

Dr. Peter L. Nagy, MD, PhD
Founder and Chief Medical Officer, Praxis Genomics
Key takeaways
- Low diagnosis rate in rare genetic diseases: Despite extensive testing, only about half of rare genetic disease cases are accurately diagnosed.
- Limited personalized cancer treatment: Just around 10% of cancer patients currently receive treatment tailored to the genetic specifics of their cancer.
- Need for enhanced genetic analysis: These statistics underscore the urgent need for improved genetic analysis and personalized treatment approaches in both rare genetic diseases and cancer.
Recording
Talk summary
Dr. Nagy started by talking about the problem of rare genetic disease: only about 50% receive a diagnosis, despite extensive testing. In addition, only about 10% of cancer patients receive personalized treatment based upon the genetic characteristics of their malignancy.
He continued with a case study (manuscript currently under review) of a 17 year old patient with a history of supravalvular stenosis (a heart valve condition), delayed development, acromelia (shortening of bones in the hands and feet) and dysmorphic facial features. Regular cytogenetic analyses (FISH and Chromosomal Microarray [CMA] and whole exome sequencing [WES]) all were negative.
The reasons CMA are limiting include relatively low resolution (10 kb events or larger), inability to detect copy-neutral variation such as inversions or translocations, and its inability to provide base-resolution breakpoints or regions of duplication.
WES is limiting as variants outside the coding region are missed; little or no CNV or structural variant information is provided; inversions or translocation information is missing; and it doesn’t detect repeat expansions or provide a mitochondrial genome assessment.
He made a powerful case for a combination of whole-genome sequencing with Optical Genome Mapping (OGM) to replace a large collection of tests and technologies: from CMA to gene panels or WES, to mtDNA sequencing and karyotyping and repeat expansion testing.
Dr. Nagy then explained his approach: to isolate high molecular weight (HMW) DNA at the outset for future OGM analyses, and start with WGS; then offer the client a ‘basic exome’ plus CNV data from exonic regions. (The rest of the data via WGS is on-hand, however only the exon-level data is provided.)
At a higher incremental price, an Expanded Exome is offered: repeat expansion data from the WGS dataset.
The next tier is Whole Genome, providing SNV data, CNVs and SVs genome-wide, in addition to an assessment of mitochondrial depletion.
The next tier is Optical Genome Mapping if balanced translocations, complex rearrangements or inversions are suspected. Lastly, for functional verification transcriptome analysis is also offered. All these test offerings do not require additional sample collection.
Dr. Nagy indicated his key technology underpinning this approach is his DNBSEQ-T7 sequencer, which they have renamed “T-Rex”, delivering 1 to 7 Tb of sequence data per day. With a four flowcell, independent architecture offering 5.8B reads per flowcell (a total of 23.2B reads per four flowcells), runs are completed in only 24 hours.
Dr. Nagy concluded with his other technologies that support Praxis Genomics, namely the Bionano Genomics Saphyr instrument for optical genome mapping, and clinical analysis reporting software for high-throughput sequencing data from a provider named Genoox.
Dr. Nagy concluded with a discussion of the individual with an undiagnosed genetic condition. He showed an IGV plot overlaid with OGM data showing a 70kb duplication on Chr7, and another plot showing a hemizygous deletion of the BRCC3 gene. There was a discrepancy between WGS and OGM data: WGS shows a deletion of a part of BRCC3 while OGM showed an insertion into BRCC3, with the insertion into BRCC3 showing a labeling pattern of the UNCX region from Chr7.
Thus this case was solved as a combination of Xq28 deletion and duplicated 7p22.3 is responsible for the phenotype. He noted in his conclusion that the combination of WGS with OGM is “likely to become recommended for genetic testing in the future”.
Investigation of herpesviruses in Alzheimer’s Disease using cerebral organoids

Dr. Elaine Lim, PhD
Assistant Professor, Department of Genomics and Computational Biology, UMass Chan Medical School
Key takeaways
- Innovative use of Stereo-Seq: Dr. Lim’s group utilized Stereo-seq, a single-cell spatial RNA sequencing technique, for advanced research in viral-human transcriptomic interactions.
- Study of cerebral organoids in genetic research: The research focused on cerebral organoids derived from the PGP1 individual of the Personal Genome Project, providing a unique model for studying genetic interactions.
- Exploration of herpes simplex virus impact: The study specifically investigated the effects of herpes simplex virus 1 (HSV-1) infection on these organoids, offering insights into viral impact on human genetics.
Recording
Talk summary
Rounding out the ASHG workshop, Dr. Lim’s group used Stereo-seq (single-cell spatial RNA sequencing) to study novel viral-human transcriptomic interactions in cerebral organoids derived from the PGP1 individual in the Personal Genome Project (who happens to be Dr. George Church, her post-doctoral mentor) and infected with herpes simplex virus 1 (HSV-1).
More than 30 years ago, Professor Ruth Itzhaki had discovered the presence of HSV-1 DNA in postmortem brains from older people and later showed evidence that HSV-1 was associated with increased risk for Alzheimer’s disease. Subsequent work by Dr. Lim’s group and her close collaborators, Drs. Benjamin Readhead and Rigel Chan, found AD-associated cellular and molecular pathologies in HSV-1 infected dissociated cells from PGP1 cerebral organoids (2D organoid cells).
Dr. Lim’s group observed interesting data where HSV infected 30-80% of the 2D organoid cells, but there was little infection of 3D intact cerebral organoids (5-15% of cells were infected). They performed single-cell spatial RNA sequencing using Stereo-seq on HSV-1 infected 3D organoids and single-cell non-spatial RNA sequencing using Parse Evercode on HSV-1 infected 2D organoid cells to compare the similarities and differences in viral-human transcript expression. Both sets of Stereo-seq and Parse libraries were sequenced on Complete Genomics’ DNBSEQ-T7 and DNBSEQ-G400 respectively.
While the 2D single-cell RNA sequence data showed significant transcriptome-wide differences, however, the 3D single-cell RNA sequence data did not. Using a statistics called GeneScore2, Dr. Lim’s team discovered that the 3D single-cell RNA sequence data had significant inter-cell cluster differences that were concordant or discordant with bulk RNA sequence data, however, the 2D single-cell RNA sequence data did not show significant inter-cell cluster differences.
These results suggest that in the 3D cerebral organoids, they could observe cell type specific tropism for HSV-1 leading to major inter-cell cluster differences in human transcript expression. However, studying the 2D organoid cells alone may not enable these discoveries made from the 3D organoids. Dr. Lim emphasized the importance of using an unbiased, transcriptome-wide, single-cell spatial RNA sequencing technology such as Stereo-seq, to gain novel insights and generate hypotheses into AD biology that cannot be discovered otherwise.
Please click here for more information about the DNBSEQ technology, click here for information about the DNBSEQ-G400, and click here to contact us with any questions.
Media Contact:
Betsy Yates
Complete Genomics
> Contact
- Gilissen C and Veltman JA et al. Genome sequencing identifies major causes of severe intellectual disability.Nature. 2014;511(7509):344-347. doi:10.1038/nature13394
- Lim ET and Church G et al. Orgo-Seq integrates single-cell and bulk transcriptomic data to identify cell type specific-driver genes associated with autism spectrum disorder. Nat Commun. 2022;13(1):3243. doi:10.1038/s41467-022-30968-3
Subscribe for exclusive content and expert insights.


















