
Decoding Cellular Diversity: Advancing Single-Cell RNA Sequencing with Complete Genomics DNBSEQ™ Technology
Feb 10, 2026
For decades, our understanding of the transcriptome was limited by the law of averages. Despite its foundational significance, conventional bulk RNA sequencing treats tissue samples as homogeneous mixtures, averaging the unique signatures of individual cells into a single profile. Biological tissues are intricate, stochastic mixtures of phenotypes. Bulk methodologies inevitably condense these dynamic populations into static averages, thereby hiding the nuances essential for understanding complex biological systems, the distinct functions of rare cell types, and the signals of subtle transcriptional variation.
The implications of this averaging are particularly severe within the context of intratumoral heterogeneity. When evaluating therapeutic response via bulk sequencing, the resulting dataset generates a consensus profile dominated by the most abundant cell populations. Consequently, if a targeted therapy successfully ablates the primary tumor clone, the aggregate data will reflect significant downregulation of the targeted pathway, suggesting clinical efficacy. However, this global view systematically fails to resolve rare, resistant subclones that function as reservoirs for disease recurrence. These minority populations, often comprising less than 1% of the tissue, are mathematically obscured by the signal of the responding majority. Thus, bulk analysis creates a diagnostic blind spot: the aggregate profile indicates molecular remission, while the occult resistant lineage persists, expands, and ultimately drives relapse.
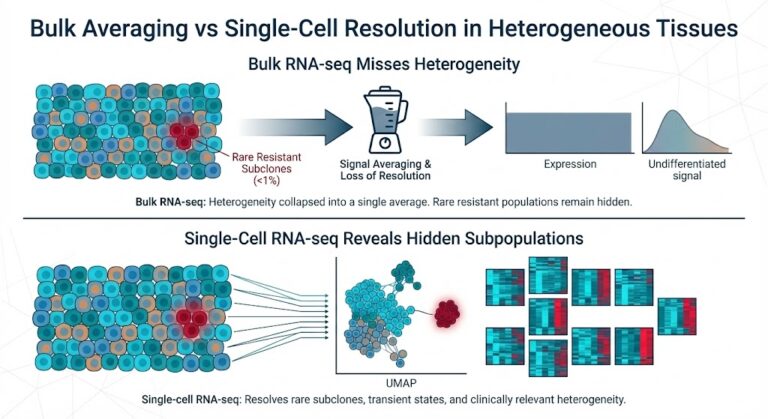
The rise of single-cell RNA sequencing (scRNA-seq) marks a significant shift, taking the field from broad, low-resolution estimates to detailed, high-definition insights. By analyzing gene expression at the level of each cell, we can finally recognize an essential truth: biological systems are built on heterogeneity. To truly understand the mechanisms driving development, pathology, and therapeutic response, we need to go beyond the consensus and identify the distinct cell types and transient states that shape the tissue microenvironment.
From rare populations to cell atlases.
Current histological standards and marker-based classifications have historically limited cellular taxonomy to broad categories defined by a handful of surface proteins. Single-cell genomics has fundamentally disrupted this classification system, necessitating the development of comprehensive reference maps, such as the Human Cell Atlas. This redefinition is driven by unsupervised clustering algorithms that categorize cells based on comprehensive transcriptomic signatures, rather than pre-selected markers, revealing that populations traditionally annotated as uniform are often identified as distinct subtypes possessing unique cytokine profiles, metabolic pathways, and spatial distributions.
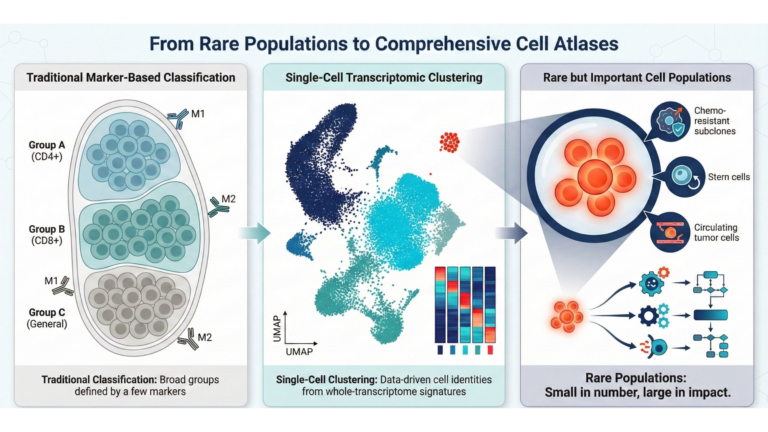
Significantly, the integrity of these new atlases depends on accurate identification of rare cell populations. In complex biological systems, the functional impact often does not correlate directly with population size; rare lineages, such as putative stem cells, circulating tumor cells, or chemo-resistant subclones, usually act as the primary drivers of disease progression despite constituting less than 1% of the tissue mass. scRNA-seq distinguishes these low-frequency events from the background noise, thereby enabling researchers to build a taxonomy that captures not only the predominant structures but also the rare populations that influence biological outcomes.
DNBSEQ Publications enabling cell atlas building.
Capturing the transcriptional state.
Although building cellular taxonomies is essential for organizing biological complexity, it is important to recognize that biological systems are inherently dynamic. Cells are not static entities characterized solely by fixed lineage markers; rather, they function as dynamic systems that perpetually adapt to environmental stimuli. scRNA sequencing effectively delineates transcriptional states, the transient functional modes that a cell adopts without altering its core identity. This distinction between cell type and cell state is critical for mechanistic understanding. A cell type represents a stable, lineage-defined identity (e.g., a CD8+ T cell), while a cell state signifies a flexible, functional condition (e.g., naïve, effector, or exhausted). By applying pseudotime analysis and RNA velocity algorithms, it is possible to reconstruct differentiation or activation trajectories, thereby inferring the directionality and rates of transcriptional changes even from a snapshot of asynchronous cells.
Many diseases are driven not by the emergence of a new cell type, but by the aberrant persistence of a specific state. In immuno-oncology, for instance, the therapeutic bottleneck is often the shift of T cells into a distinct exhausted transcriptional program characterized by the upregulation of inhibitory receptors. Identifying the specific regulatory networks that lock cells into these dysfunctional states enables the development of modulators that can effectively rewire the cell back to a functional phenotype. This intervention is impossible to design without the resolution to define the state itself.
DNBSEQ Publications enabling transcriptional studies.
Capturing Biological Complexity with DNBSEQ technology.
The key shift from bulk averaging to single-cell resolution represents a crucial step toward understanding biological complexity. By revealing hidden taxonomies, identifying dynamic transcriptional states, and isolating rare, high-impact cell populations, scRNA-seq has become an essential tool for mechanistic discovery. However, successfully achieving these goals, moving from small-scale hypothesis testing to building statistically robust, large-scale biological atlas building, is mainly limited by sequencing throughput and costs.
Complete Genomics DNBSEQ-T1+ and DNBSEQ-T7 sequencing platforms combine high throughput, exceptional accuracy, and cost efficiency to meet the demands of large-scale single-cell research. By leveraging DNBSEQ™ technology to minimize index hopping and reduce amplification bias, researchers can identify rare cell populations with greater confidence than with traditional sequencing methods. This high-throughput, low-error approach is crucial in constructing new taxonomies for the Human Cell Atlas or delineating the dynamic cellular states within the tumor microenvironment.
The future of precision medicine and foundational biology depends on accurately capturing cellular heterogeneity. Complete Genomics’ technologies are specifically designed to enable this next generation of genomic insights. By providing the data density and precision necessary for large-scale single-cell analysis, Complete Genomics helps transform cellular heterogeneity from a diagnostic challenge into a therapeutic opportunity. While single-cell RNA sequencing offers an unprecedented view of cellular identity and transcriptional states, it does so by dissociating cells from their native tissue context. Stereo-seq spatial transcriptomics technology extends this resolution by mapping gene expression directly within intact tissues, preserving the architecture and cellular interactions that influence development, disease progression, and therapeutic response. Collectively, single-cell and spatial transcriptomics establish a comprehensive framework for understanding biological complexity across both molecular and tissue levels.
Explore how Complete Genomics advances spatial transcriptomics
Subscribe for exclusive content and expert insights.







